Das nephrotische Syndrom ist durch schwere Proteinurie, Hypoalbuminämie und periphere Ödeme gekennzeichnet. Im Gegensatz dazu geht das nephritische Syndrom mit Hämaturie, variablem Verlust der Nierenfunktion und Bluthochdruck einher, wobei es hier allerdings auch zu Überschneidungen der Symptome kommen kann. Die primären Ätiologien des nephrotischen Syndroms sind die Minimal-Change-Glomerulonephritis Minimal-Change-Glomerulonephritis Minimal-Change-Glomerulonephritis, die membranöse Glomerulonephritis Glomerulonephritis Membranoproliferative Glomerulonephritis und die fokal segmentale Glomerulosklerose. Das klinische Erscheinungsbild des nephrotischen Syndroms umfasst eine Proteinurie (> 3,5 g/Tag), eine Hypalbuminämie (< 3 g/dl) und periphere Ödeme. Außerdem tritt häufig eine Hyperkoagulabilität Hyperkoagulabilität Hyperkoagulopathien aufgrund des Proteinverlustes auf, sodass es im Verlauf häufiger zu thromboembolischen Komplikationen kommen kann. Die Diagnose kann klinisch erfolgen, in den meisten Fällen ist aber eine Nierenbiopsie erforderlich. Die Behandlung variiert je nach Ätiologie und umfasst in der Regel Kortikosteroide oder andere immunsuppressive Medikamente.
Kostenloser
Download
Lernleitfaden
Medizin ➜
Das nephrotische Syndrom ist gekennzeichnet durch eine schwere Proteinurie (> 3,5 g/Tag), niedriges Serumalbumin (< 3 g/dl) und periphere Ödeme. Die typischen Symptome entwickeln sich, wenn der renale Eiweißverlust die Albuminsyntheseleistung der Leber Leber Leber überschreitet.
Hauptsächlich beteiligt an der Genese des primäres nephrotisches Syndrom sind die Podozyten. Eine erhöhte Filtration durch die glomeruläre Kapillarwand führt zu Proteinurie.
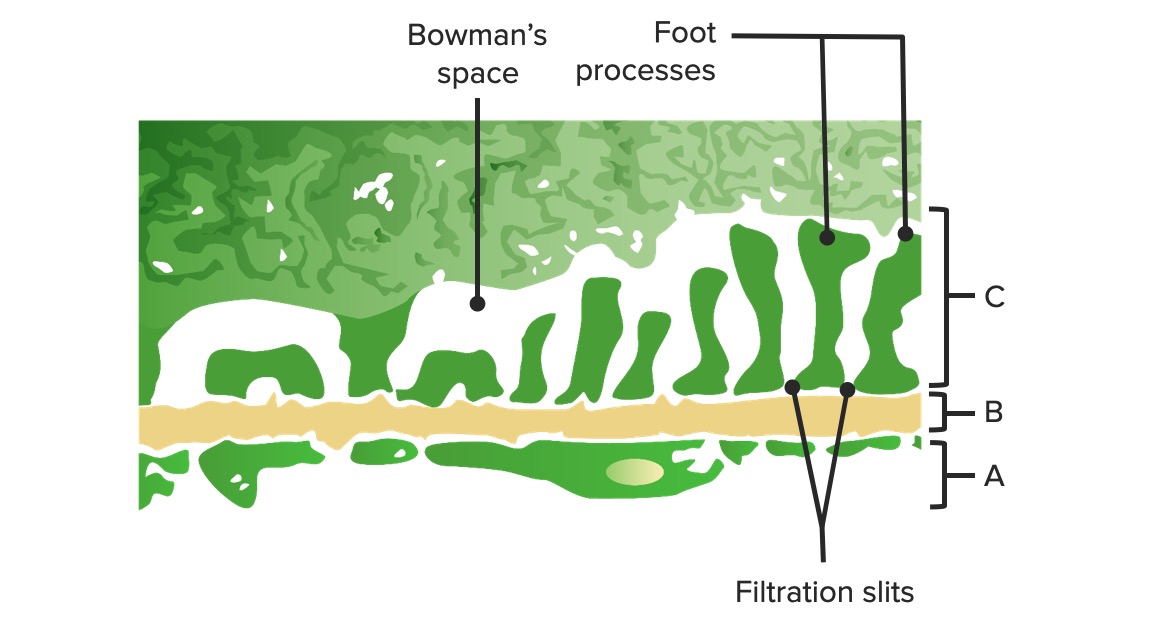
Schema der glomerulären Barriere:
A: Fenestriertes Endothel der glomerulären Kapillaren
B: Glomeruläre Basalmembran
C: Epithelschicht, welche aus Podozytenfortsätzen und Strukturproteinen gebildet wird und das Schlitzdiaphragma bildet
Bild von Lecturio.
Primäre Ätiologien treten eher mit den klassischen Befunden eines nephrotischen Syndroms (Proteinurie, Ödem, Hypalbuminämie) auf, während sekundäre Ursachen eher mit einer Proteinurie im nephrotischen Bereich auftreten. Die Diagnose wird durch die klinische Anamnese und Präsentation, den Grad der Proteinurie und den Biopsiebefund gestellt.

Ovale Fettkörper bei der Urinuntersuchung
Bild : „Unidentified structures in urine“ von Ed Uthman. Lizenz: CC BY 2.0
Elektronenmikroskopie zeigt glomeruläre Kapillarschleifen mit Befunden einer Minimal-Change-GN:
Podozyten mit ausgedehnter, diffuser Auslöschung des Fußfortsatzes (weiße Pfeile) und mikrovillöser Transformation
Keine elektronendichten Ablagerungen
Normale Dicke der glomerulären Basalmembran.

Person mit sowohl membranöser Nephropathie als auch diabetischer Nephropathie:
A: Ablagerungen von IgG in der Basalmembran erscheinen als diffuses granuläres Muster, wie in der Immunfluoreszenz (×200) gezeigt.
B: Lichtmikroskopie zeigt membranöse Glomerulonephritis mit verdickten und hervortretenden Kapillarschleifen. Zahlreiche körnige und dichte Ablagerungen in den subepithelialen Bereichen lokalisiert (PAS-Färbung, ×200).
C: Elektronenmikroskopische Aufnahme einer verdickten glomerulären Basalmembran (GBM) mit zahlreichen körnigen, dichten Ablagerungen in subepithelialen Bereichen (×5000).
Die folgenden Ätiologien treten mit geringerer Wahrscheinlichkeit beim klassischen nephrotischen Syndrom auf, können jedoch immer noch bei Nierenbiopsien in Fällen mit Proteinurie im nephrotischen Bereich gefunden werden.
Diabetische Nephropathie Diabetische Nephropathie Chronische Komplikationen bei Diabetes mellitus:
Membranoproliferative Glomerulonephritis Glomerulonephritis Membranoproliferative Glomerulonephritis (MPGN):
Amyloidose Amyloidose Amyloidose:

Diabetische Nephropathie:
(a): Lichtmikroskopie mit HE-Färbung zeigt eine ausgedehnte mesangiale Expansion ohne eine deutliche Zunahme der Zellularität. Eine Kimmelstiel-Wilson (KW)-Läsion ist hier dargestellt und bezieht sich auf die noduläre Glomerulosklerose, die in der Späterkrankung zu sehen ist, aber nicht so häufig ist wie die diffuse diabetische Glomerulosklerose. Kimmelstiel-Wilson-Läsionen sind normalerweise kugelförmig und eosinophil und haben einen zentralen hypozellulären oder azellulären Bereich. Mesangiale Expansion und KW-Läsionen sind beide auf eine erhöhte Produktion extrazellulärer Matrix zurückzuführen.
(b): Elektronenmikroskopie zeigt eine verdickte Basalmembran und eine Auslöschung des Fußfortsatzes von Podozyten.

Membranoproliferative Glomerulonephritis:
PAS-positive „hyaline Thromben” innerhalb der Kapillarlumina (PAS-Färbung, Bildvergrößerung × 400)

Amyloidablagerungen in den 3 großen Kompartimenten der Niere:
A: Glomeruläre Amyloidablagerungen überwiegend in Mesangialräumen (HE, x400, Pfeil).
B: Interstitielle Amyloidose (HE, x200).
C: Gefäßamyloidose (HE, x100).
D: Kongorotfärbung von vaskulären Amyloidablagerungen (x100).



















